


Teilnahme an klinischen Studien
Erstellung der Leitlinie
1Allgemeine Informationen
Klinische Studien werden mit dem Ziel durchgeführt, aktuelle Behandlungsmöglichkeiten einer Erkrankung zum Nutzen der Patient*innen weiter zu verbessern. Die Teilnahme an solchen Studien eröffnet Ihnen die Chance, frühzeitig mit modernen und neuen, vielleicht effektiveren Therapien behandelt zu werden. Mit Ihrer Teilnahme leisten Sie zudem einen wichtigen Beitrag zur weiteren Optimierung der bestehenden Therapiekonzepte. Während Ihrer Teilnahme an der Studie werden Sie intensiv überwacht und untersucht. Klinische Studien können auch mit zusätzlichen Belastungen verbunden sein, da Sie eventuell regelmäßig Termine wahrnehmen müssen, die zur Verlaufsbeobachtung über die Behandlung hinaus erforderlich sind. Je nach Studie werden darüber hinaus oft auch Patientenbefragungen durchgeführt, so dass neben dem medizinischen Aspekt (Behandlungserfolg) auch der psychische bzw. psychoonkologische Aspekt betrachtet wird (Lebensumstände / Lebensqualität etc.). Zudem kann die untersuchte Behandlung dem sonst üblichen Therapiestandard über- oder unterlegen sein. Dies herauszufinden ist ein wichtiger Aspekt klinischer Studien.
1.1Wichtige Bestandteile einer Studie
1.1.1Das Protokoll
Die Planung einer klinischen Studie beginnt in der Regel mit der Erstellung eines Studienprotokolls, das auch als „Prüfplan“ bezeichnet wird. In dem Studienprotokoll werden der klinische Ablauf der Studie beschrieben, die Studienendpunkte („Ziele“), die Methode zur Datenauswertung, die zu erhebenden Messwerte (Laborwerte, körperliche Untersuchungen etc.) sowie die Ein- und Ausschlusskriterien genau festgelegt. Dies garantiert eine vergleichbare, sichere Behandlung aller in der Studie behandelter Patient*innen und ermöglicht nach Abschluss der Studie eine sichere statistische Auswertung der Behandlungsergebnisse.
1.1.1.1Ziele der Studie
Die Ziele einer klinischen Studie werden auch als „Endpunkte“ der Studie bezeichnet. Es gibt primäre und sekundäre Endpunkte in Studien. Der primäre Endpunkt ist das vorher festgelegte, vorrangige Ziel einer klinischen Studie. Bei großen Studien ist dies oft die Verbesserung der Überlebenschancen. Aber auch das Ansprechen auf die Behandlung oder die Verminderung von Nebenwirkungen kann ein primärer Endpunkt sein. Der sekundäre Endpunkt beschreibt hingegen nachrangige Ziele einer klinischen Studie. Dieser nachrangige Endpunkt kann den Therapieeffekt oder die Sicherheit einer Therapie zusätzlich beschreiben, ist aber allein kein Beweis für die Wirksamkeit oder Überlegenheit einer neuen Therapie.
1.1.1.2Ein- und Ausschlusskriterien
Diese Kriterien beinhalten meist Bedingungen, die eine sichere Durchführung der Studie gewährleisten. Nur wenn ein Patient alle Einschlusskriterien erfüllt, keine Ausschlusskriterien vorliegen und sein schriftliches Einverständnis zur Studienteilnahme vorliegt, kann eine Aufnahme des Patient*innen in die Studie erfolgen.
1.1.2Die Patienteninformation
Jeder Patient, der innerhalb einer Studie behandelt wird, erhält vor Einschluss in die Studie eine umfassende Aufklärung durch die behandelnden Ärzt*innen. Teil dieser Aufklärung ist die Patienteninformation, die Ihnen in schriftlicher Form ausgehändigt wird. Dieses Schriftstück enthält alle für Patient*innen wichtigen Informationen, um den Umfang der Studie und die damit verbundenen Risiken abschätzen zu können. So sind die im Rahmen der Studie geplante Behandlung, deren Ablauf, die Untersuchungen innerhalb der Studie, Nebenwirkungen der Therapie sowie rechtliche Informationen zur Versicherung und dem Datenschutz wichtige Bestandteile der Patienteninformation. Die Rechte und Pflichten im Zusammenhang mit der Studie werden ebenfalls beschrieben. Inhaltlich kann sich die Patienteninformation von Studie zu Studie unterscheiden.
TIPP: Nach der Aushändigung der Patienteninformation und der Aufklärung durch Ihren Arzt müssen Sie sich nicht sofort für oder gegen eine Studienteilnahme entscheiden. Üblich ist das Einräumen einer Bedenkzeit von mindestens 24 Stunden.
1.1.3Die Ethikkommission
Vor dem Einschluss der ersten Patient*innen in eine Studie muss zudem die Zustimmung von der zuständigen Ethikkommission (sogenanntes Ethikvotum) eingeholt werden. Die Ethikkommission ist ein unabhängiges und übergeordnetes Kontrollorgan. In jedem Bundesland gibt es mindestens eine Ethikkommission. Ihre Mitglieder gehören verschiedenen Berufsgruppen an. Meist sind es Mediziner, Juristen, Naturwissenschaftler, Theologen oder Philosophen und Laien. Die Zusammensetzung wird durch das Landesrecht bestimmt. Die jeweilige Ethikkommission überprüft, ob alle Schutzbestimmung zur Durchführung von Studien eingehalten werden und ob zum Bespiel der mögliche Nutzen der Studie mögliche Risiken überwiegt. Auch der Inhalt der Patienteninformation ist Gegenstand der Überprüfung.
1.1.4Datenauswertung
Während der Therapie werden regelmäßig die Daten zur Patientensicherheit und Behandlungswirksamkeit während einer laufenden klinischen Studie durch eine unabhängige Expertengruppe überwacht. Diese Gruppe wird als Datenüberwachungskomitee (Data Monitoring Committee oder Data Safety Monitoring Board) bezeichnet. Nach Abschluss der Therapie werden die erhobenen Daten und Messwerte mithilfe von statistischen Analysemethoden im Hinblick auf die primären und sekundären Endpunkte ausgewertet. Diese Analysen werden durch erfahrene Biostatistiker betreut. Nach Abschluss der Datenauswertung werden die Studienergebnisse in der Regel in der medizinischen Fachpresse veröffentlicht.
1.2Ablauf einer klinischen Studie
In der nachfolgenden Abbildung sind die einzelnen Bestandteile des Ablaufs der Teilnahme an einer klinischen Studie zur besseren Übersicht nochmal dargestellt.
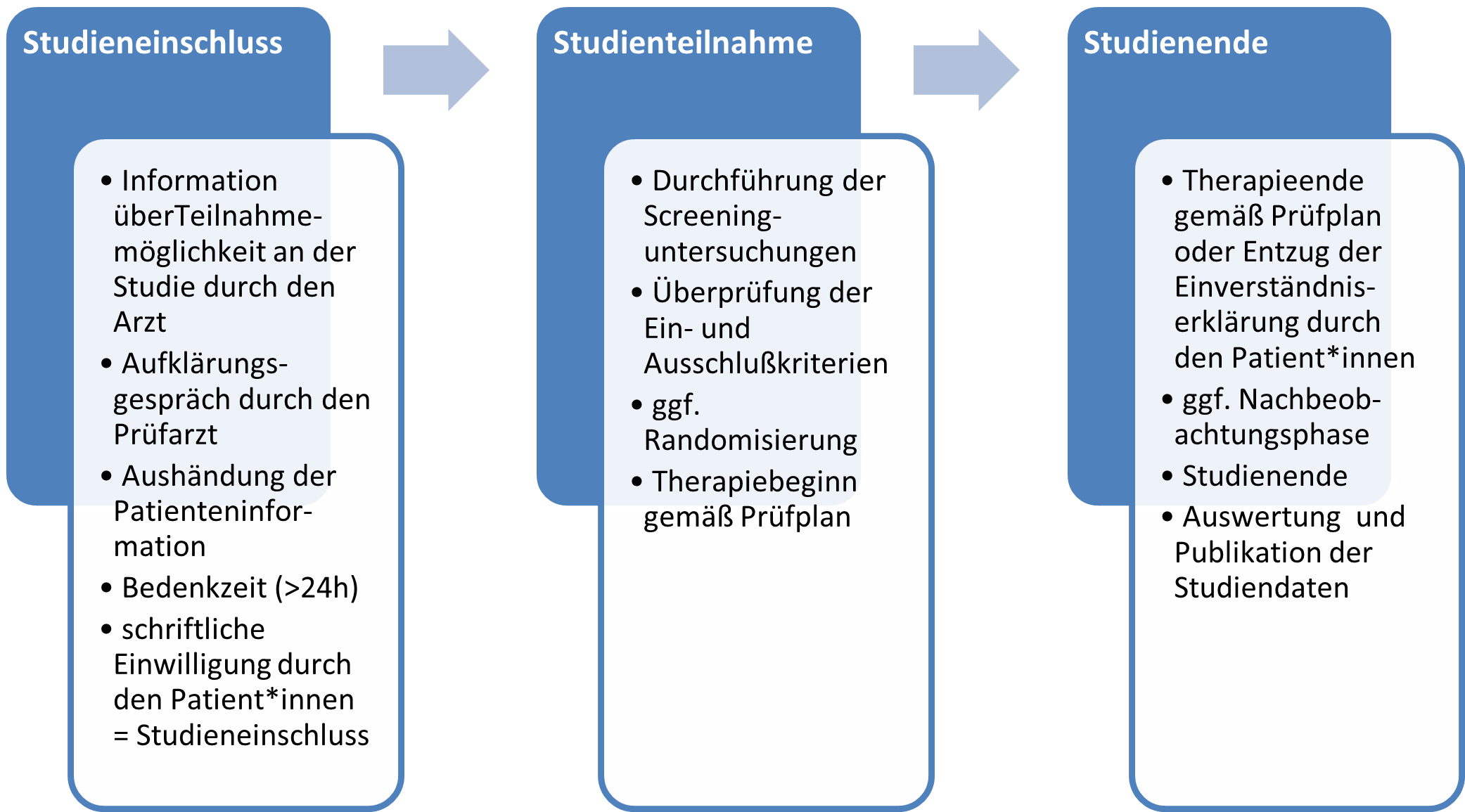
1.3Studienarten
Es wird zwischen interventionellen (Kapitel 1.3.1) und nicht-interventionellen (Kapitel 1.3.2) Studien unterschieden. Während es sich bei nicht-interventionellen Studien häufig um Beobachtungsstudien handelt, wird bei Interventionsstudien eine Behandlung als geplante und gezielte Maßnahme (Intervention) aktiv durchgeführt.
Nachfolgend möchten wir Ihnen einen kurzen Überblick über die unterschiedlichen Arten von klinischen Studien geben.
1.3.1Interventionelle Studien
Es gibt zwei Formen von Interventionsstudien: die randomisierte und die nicht-randomisierte kontrollierte Studie. In der randomisierten Form werden die Patient*innen in Gruppen aufgeteilt und zufällig in einem zuvor festgelegten Verhältnis zu den einzelnen zu vergleichenden Therapien zugeteilt. Wird bspw. in einer zweiarmigen randomisierten Studie im Verhältnis 1:1 randomisiert oder zugeteilt, so bedeutet dies, dass die gleiche Anzahl Patient*innen in beide Gruppen (Arme) nach dem Zufallsprinzip zugeteilt werden. Diese Zuteilung ist nicht durch Arzt oder Patient beeinflussbar.
Klinische Interventionsstudien müssen vom Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) oder vom Paul-Ehrlich-Institut (PEI) zugelassen werden. Man unterscheidet bei diesen Studien vier Phasen:
1.3.1.1Phase I-Studie
In der Phase I der klinischen Prüfung wird die Studie an einer sehr kleinen Anzahl gesunder Studienteilnehmer durchgeführt, um die Verträglichkeit und Sicherheit des neuen Arzneimittels zu bestimmen.
1.3.1.2Phase II-Studie
Basierend auf den Ergebnissen der Phase I-Studie wird anschließend eine Phase II-Studie an einem kleinen Patientenkollektiv durchgeführt, um die optimale Dosierung des Medikaments festzulegen und erste Daten zur Wirksamkeit und Verträglichkeit des Medikaments an Patient*innen zu erheben.
1.3.1.3Phase III-Studie
Im nächsten Schritt wird die Phase III-Studie an einem großen Patientenkollektiv durchgeführt, um die Wirksamkeit des neuen Therapieverfahrens nachzuweisen. Phase III-Studien werden manchmal auch als Zulassungsstudien bezeichnet, da die Ergebnisse der Studie darüber entscheiden können, ob ein neues Medikament die Zulassung (vom BfArM) zur Therapie der untersuchten Erkrankung erhält.
Häufig werden Phase III-Studien als randomisierte kontrollierte Studien beziehungsweise Vergleichsstudien durchgeführt. Das bedeutet, dass bei Studieneinschluss die Studienteilnehmer in unterschiedliche Gruppen aufgeteilt werden und nach Vollendung der Studie die Behandlungs-ergebnisse der unterschiedlichen Gruppen miteinander verglichen werden. Während zum Beispiel die eine Gruppe die bisherige Standardtherapie erhält und die Kontrollgruppe bildet, wird die andere Gruppe mit dem neuen Therapieansatz behandelt und bildet die Behandlungsgruppe. Die Verteilung der Studienteilnehmer auf die einzelnen Behandlungsgruppen erfolgt per Zufallsprinzip (Randomisierung). Weder Ärzt*innen noch Patient*innen haben einen Einfluss auf die Zuteilung in die jeweilige Behandlungsgruppe der Studie. Wichtig ist bei derartigen Studien, dass zu Beginn nicht bekannt ist, welcher Therapieansatz der bessere ist. Sobald sich eine bessere/schlechtere Wirkung herausstellt, werden solche Studien gestoppt und alle Studienpatient*innen erhalten dann die bessere Therapie.
1.3.1.4Phase IV-Studie
In Phase IV-Studien erhalten die Studienteilnehmer*innen ein bereits zugelassenes Medikament. Sie werden beispielsweise durchgeführt, um die Wirksamkeit des Medikaments nochmal in einer größeren Patientengruppe unter alltäglichen Bedingungen gezielt zu untersuchen oder seltene Nebenwirkungen zu erheben. Dies ist manchmal wichtig, da in den Zulassungsstudien die Verabreichung des Medikamentes auf einen bestimmten Personenkreis eingeschränkt ist (z.B. sind ältere und kränkere Patient*innen oft ausgeschlossen).
1.3.1.5Therapieoptimierungsstudien
Therapieoptimierungsstudien zielen darauf ab, eine bereits eingesetzte Therapie weiter zu verbessern, um die Heilungsaussichten zu steigern, die Lebensqualität der Patient*innen zu verbessern oder auch um Langzeitfolgen der Krebstherapie zu reduzieren. Therapieoptimierungsstudien werden oft von Studiengruppen durchgeführt, die zum Beispiel den in Kapitel 3 aufgeführten Kompetenznetzen angehören.
1.3.2Nicht-interventionelle Studien
In einigen Situationen sind kontrollierte und randomisierte Studien nicht möglich, weil die Voraussetzungen für diese Form der Untersuchung nicht gegeben sind. Die Gründe hierfür können bspw. sein, dass es zu wenige Patient*innen mit einer bestimmten Erkrankung gibt, so dass die Gruppe an Patient*innen zu klein wäre, um eine sichere Aussage zum Nutzen der Therapie in einer Studie gewinnen zu können. Ein weiterer Grund können ethische Gründe darstellen. Beispielsweise, wenn eine Gruppe in einer randomisierten Studie einen offensichtlichen Nachteil in der Behandlung hätte oder zum Vergleich eine gewebsverletzende (invasive) Therapie wie eine Operation erforderlich wäre. Nicht-interventionelle Studien bieten dem Studienteilnehmer selbst keinen unmittelbaren Vorteil. Sie können jedoch dazu beitragen, bestehende Therapiekonzepte weiter zu optimieren.
Zu den nicht-interventionellen Studien zählen Querschnitts- und Kohortenstudien ebenso wie Fall-Kontrollstudien:
1.3.2.1Querschnittsstudie
Querschnittsstudien sind Momentaufnahmen und beschreiben die Verteilung z.B. einer Erkrankung in der Bevölkerung zu einem bestimmten Zeitpunkt.
1.3.2.2Kohortenstudie
In einer Kohortenstudie werden Patientengruppen (Kohorte), die eine bestimmte Therapie (Intervention) erhalten haben, über einen längeren Zeitraum nachbeobachtet und hinsichtlich des Therapieansprechens nachverfolgt. Es kann sich dabei sowohl um vorausschauende (prospektive) als auch rückblickende (retrospektive) Studien handeln.
1.3.2.3Fall-Kontroll-Studie
Werden die Ergebnisse des Behandlungserfolgs rückblickend (retrospektiv) erhoben und ausgewertet, ist häufig auch von Fall-Kontroll-Studien die Rede. Sie ermöglichen auch einen Vergleich zwischen vorher definierten Gruppen, z.B. hinsichtlich des Vorliegens von Risikofaktoren in einer Gruppe von Patient*innen mit einer bestimmten Erkrankung („Fälle“) und einer Gruppe von Patient*innen ohne diese Erkrankung („Kontrollen“).
1.3.2.3.1Gut zu wissen
Welche Rechte und Pflichten habe ich als Teilnehmer an einer klinischen Studie?
Patient*innen, die gut informiert sind und ihre Rechte kennen, können den Ärzt*innen, der Krankenkasse oder auch dem Apotheker als gleichberechtigter Partner gegenübertreten.
Bevor Sie Ihre schriftliche Einwilligung zur Teilnahme an einer klinischen Studie geben, haben Sie das Recht, über den Inhalt und Zweck, den Ablauf sowie Risiken und Vorteile der klinischen Studie umfassend aufgeklärt zu werden. Dazu erhalten Sie sowohl ein Patienteninformationsformular als auch ein Aufklärungsgespräch mit dem Arzt. Wenn Sie sich zur Teilnahme an einer klinischen Studie entscheiden, dann sollten Sie die Empfehlungen des Arztes sowie die in der Studie vorgeschriebenen Untersuchungszeitpunkte einhalten. Ihre Zuverlässigkeit als Teilnehmer*in ist für die Aussagekraft und den erfolgreichen Abschluss der Studie von besonderer Bedeutung. Wenn Sie Fragen zu den Wirkungen und Nebenwirkungen der Therapie haben oder diese während der Therapie bemerken, besprechen Sie auch diese bitte immer mit den Studienärzt*innen. Sie haben das Recht, die Teilnahme an einer Studie jederzeit abzubrechen. Sollten sich während Ihrer Teilnahme an der Studie wissenswerte Neuigkeiten innerhalb der Studie ergeben, so werden Sie auch darüber informiert.
Entstehen mir Nachteile, wenn ich meine Zusage zur Teilnahme an der Studie zurückziehe?
Die Studienteilnahme erfolgt grundsätzlich auf freiwilliger Basis. Wird die Einwilligung zur Teilnahme an der Studie zurückgezogen, werden Ihre Ärzt*innen Ihnen die beste verfügbare Standardtherapie vorschlagen. Von potentiellen Vorteilen des innerhalb der Studie eingesetzten Medikaments (sogenanntes Prüfpräparat) oder diagnostischer Methoden können Sie dann gegebenenfalls nicht mehr profitieren, da die Studienmedikation nur mit Ihrem Einverständnis angewendet werden darf.
Was ist eine Doppelblind-Studie?
Bei einer Doppelblind-Studie werden unterschiedliche Gruppen (Kontrollgruppe, Versuchsgruppe) miteinander verglichen. Die Zuteilung des Studienteilnehmers in eine der Gruppen erfolgt per Zufallsprinzip. Weder die Studienteilnehmer*in noch die Studienärzt*innen können Einfluss auf die Zuteilung zur jeweiligen Studiengruppe nehmen. Auch haben weder Ärzt*innen noch Patient*innen Kenntnis darüber, in welcher Gruppe sich die Studienteilnehmer*innen befindet.
Was passiert mit meinen Daten?
Im Rahmen klinischer Studien werden Patient*innen über die Verwendung Ihrer Daten während des Aufklärungsgespräches unterrichtet. Die in der Studie gesammelten Daten werden je nach Studienart und Ziel der Studie verwendet. Häufig werden diese Daten „pseudonymisiert“, d.h. verschlüsselt und ohne Namensnennung oder sonstige persönliche Daten verarbeitet, so dass kein Rückschluss auf Ihre Person möglich ist. Je nach Studie enthält die Patienteninformation genaue Angaben darüber, in welcher Form die gesammelten Daten für welchen Zeitraum an welche Institution gemeldet werden. Der Umfang dieser Datenübermittlung, sowie der Zeitraum der Nutzung Ihrer Daten wird ebenfalls für die jeweilige Studie in der Patienteninformation genannt. Sie können jederzeit Auskunft über Ihre gespeicherten Daten verlangen. Sie haben zudem das Recht, fehlerhafte personenbezogene Daten berichtigen oder löschen zu lassen und zu jeder Zeit die Einwilligung zur Verarbeitung ihrer personenbezogenen Daten zu widerrufen. Im Falle einer solchen Rücknahme Ihrer Einwilligung dürfen, die bis zu diesem Zeitpunkt gespeicherten Daten weiterhin verwendet werden, soweit dies nach Arzneimittelgesetz erforderlich ist. Das Arzneimittelgesetz enthält nähere Vorgaben für den erforderlichen Umfang der Einwilligung in die Datenerhebung und -verwendung.
Bin ich als Teilnehmer einer klinischen Phase I-IV Studie versichert?
Für die Dauer der Behandlung sind Patient*innen in klinischen Studien versichert. Umfang der Versicherung und Dauer des Versicherungsschutzes kann jedoch für jede Studie individuell sein. Daher sind genaue Informationen zu den jeweiligen Patientenversicherungen in der Patienteninformation zusammengestellt. Dort wird der Versicherungsschutz im Detail beschrieben.
2Tipps und Tricks
Bereiten Sie sich auf das Aufklärungsgespräch mit dem Studienarzt vor!
Lesen Sie die Patienteninformation sorgfältig durch, markieren Sie sich die Textstellen, zu denen Sie Fragen haben und / oder notieren Sie sich die Fragen und stellen Sie diese dem Studienarzt während des Aufklärungsgespräches. Zudem ist es hilfreich, einen Angehörigen oder Freund zum Aufklärungsgespräch mitzunehmen.
Lassen Sie sich die einzelnen Behandlungsschritte genau erklären und fragen Sie auch, ob es andere Möglichkeiten dazu gibt. Wenn Sie etwas nicht verstanden haben, fragen Sie nach, bis Ihnen alles klar ist. Sollten Sie Zweifel haben oder eine Bestätigung suchen, holen Sie von einem anderen Arzt oder in einer anderen Klinik eine zweite Meinung ein.
Erkundigen Sie sich nach Möglichkeiten zum Fruchtbarkeitserhalt!
Krebs oder die Therapie der Erkrankung können in einigen Fällen die Fruchtbarkeit schädigen. Daher gilt auch im Rahmen von Studien, dass bei bestehendem Kinderwunsch vor Start der Therapie die Möglichkeiten zum Fruchtbarkeitserhalt besprochen werden sollen, um gegebenenfalls eine Option zur Fruchtbarkeitserhaltung wahrnehmen zu können. In der AYAPedia Leitlinie „Fruchtbarkeit und Fruchtbarkeitserhalt“ haben wir weitere Informationen für Sie zusammengestellt.
Fragen Sie den Studienarzt, ob im Rahmen der Studie eine Aufwandsentschädigung vorgesehen ist.
Reisekosten, die im Zusammenhang mit der Teilnahme an einer Studie entstehen, werden manchmal erstattet. Es kann sich also lohnen, gezielt danach zu fragen.
Suchen Sie auch während der Studie das Gespräch mit Ihren behandelnden Ärzt*innenn, falls Fragen oder Unsicherheiten entstehen!
Häufig lässt der Klinikalltag für Ihre behandelnden Ärzt*innen keine langen Patientengespräche zu. Die Termine sind eng getaktet, die Ärzt*innen fassen sich oft kurz oder/und wirken gestresst. Dies kann dazu führen, dass Sie sich nicht trauen, notwendige Fragen zu stellen. Denken Sie daran, dass Ihnen jederzeit das Recht zusteht, alle Ihnen wichtigen Fragen zu klären. Es hilft, wenn Sie sich vor dem Termin die offenen Fragen notieren und im Gespräch die Notizen als Gedächtnisstütze nehmen.
Eine weitere Möglichkeit bieten die Studienzentralen der Forschungsgruppen, die Studien planen und durchführen. Viele dieser Gruppen bieten die Option der Kontaktaufnahme und Unterstützung bei offenen Fragen zur Erkrankung und den Therapieoptionen. Kontaktadressen finden sich im nachfolgenden Kapitel 3 (weiterführende Informationen).
3Weiterführende Informationen
Das Deutsche Register klinischer Studien bietet Informationen über alle klinischen Studien in Deutschland: https://www.drks.de/drks_web/
Blauer Ratgeber der Krebshilfe zu Klinischen Studien: https://www.krebshilfe.de/informieren/ueber-krebs/infothek/infomaterial-kategorie/die-blauen-ratgeber/
Service des Ärztlichen Zentrums für Qualität in der Medizin (ÄQZ) mit Informationen zu klinischen Studien: https://www.patienten-information.de/kurzinformationen/klinische-studien
Aufklärung über Patientenrechte: https://www.patientenbeauftragter.de/patientenrechte/
Kompetenznetz Maligne Lymphome: https://lymphome.de/studien/
Kompetenznetz Leukämien: https://www.kompetenznetz-leukaemie.de/content/studien/
Europäische Patientenakademie – Das ABC der Arzneimittelentwicklung: https://www.eupati.eu/de/
5Erklärungen zu möglichen Interessenkonflikten
nach den Regeln der tragenden Fachgesellschaften.
Download
Reference:
Quellenangabe:
Onkopedia-Leitlinien werden kontinuierlich an den Stand des Wissens angepasst. Die jeweils gültige Version, AGB und Nutzungsbedingungen finden Sie unter www.onkopedia.com.
Für die kommerzielle Nutzung wenden Sie sich bitte an onkopedia@dgho.de.
Onkopedia guidelines are continuously adapted to the state of knowledge. The currently valid version, terms of use and general terms and conditions can be found at onkopedia-guidelines.info.
For commercial use, please contact onkopedia@dgho.de.
